


Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent
Title: Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent
Authors: Koki Fujimori, Mitsuru Ishikawa, Asako Otomo, Naoki Atsuta, Ryoichi Nakamura, Tetsuya Akiyama, Shinji Hadano, Masashi Aoki, Hideyuki Saya, Gen Sobue & Hideyuki Okano
Journal: Nature Medicine, 24, pages1579-1589(2018)
DOI: 10.1038/s41591-018-0140-5
Abstract:
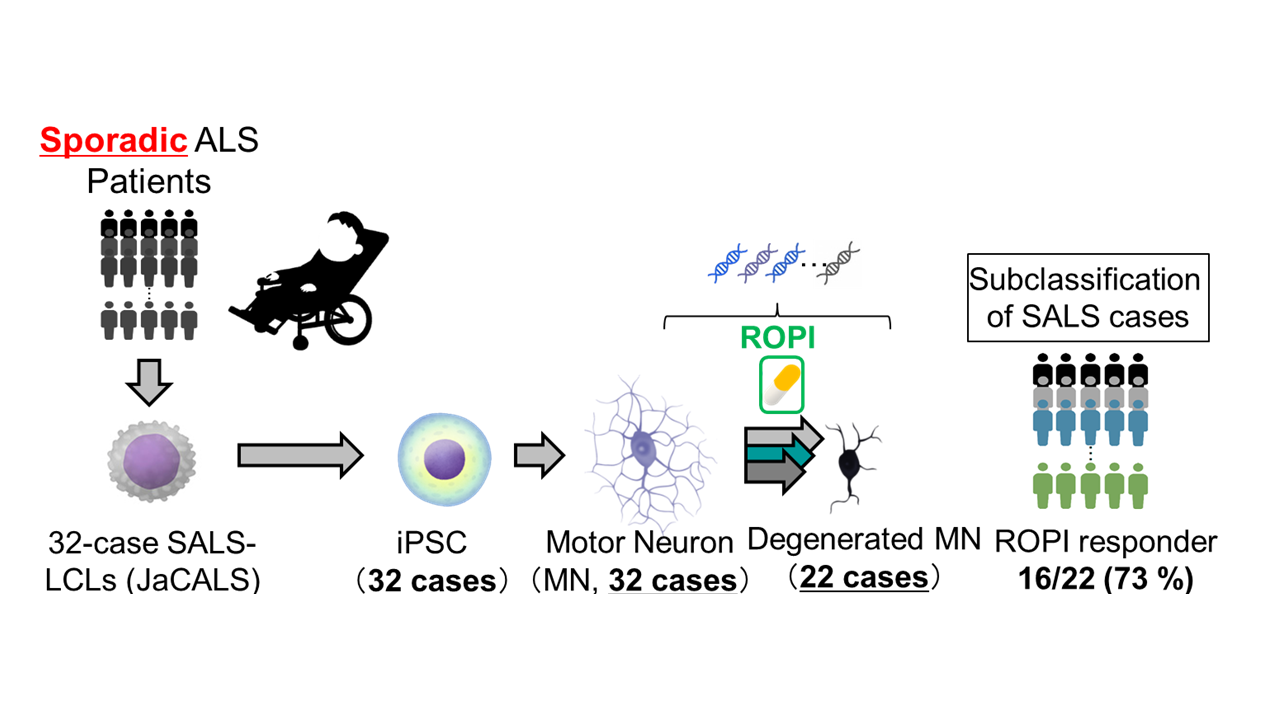
Amyotrophic lateral sclerosis (ALS) is a heterogeneous motor neuron disease for which no effective treatment is available, despite decades of research into SOD1-mutant familial ALS (FALS). The majority of ALS patients have no familial history, making the modeling of sporadic ALS (SALS) essential to the development of ALS therapeutics. However, as mutations underlying SALS pathogenesis have not yet been identified, it remains difficult to establish useful models of SALS. Using induced pluripotent stem cell (iPSC) technology to generate stem and differentiated cells retaining the patients' full genetic information, we have established a large number of in vitro cellular models of SALS. These models showed phenotypic differences in their pattern of neuronal degeneration, types of abnormal protein aggregates, cell death mechanisms, and onset and progression of these phenotypes in vitro among cases. We therefore developed a system for case clustering capable of subdividing these heterogeneous SALS models by their in vitro characteristics. We further evaluated multiple-phenotype rescue of these subclassified SALS models using a FDA-approved drug (ropinirole) which had been selected based on the effect for non-SOD1 FALS models. Integration of the datasets acquired in this study permitted the visualization of molecular pathologies shared across a wide range of SALS models.
Links:
今月のサイエンス